Our latest release of intOGen now available online!
We are very excited to release a new version of IntOGen, our framework to identify cancer genes and pinpoint putative mechanism of tumorigenesis. The IntOGen website [...]
We are very excited to release a new version of IntOGen, our framework to identify cancer genes and pinpoint putative mechanism of tumorigenesis. The IntOGen website [...]
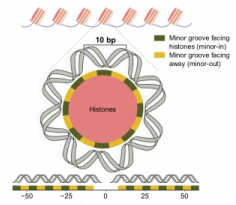
We are very excited to share that our work describing the somatic and germline 10bp periodicity in the mutation rate within nucleosome-bound DNA was published in the [...]
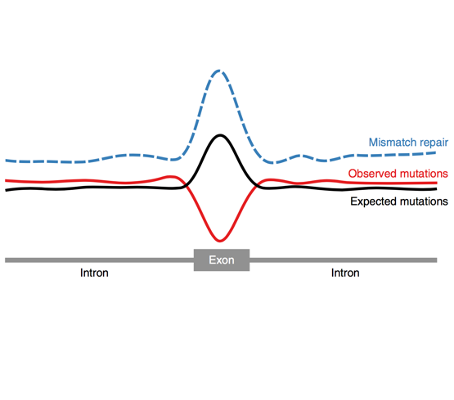
We are happy to announce that our paper on reduced mutation burden in exons caused by differential mismatch repair has been published yesterday, 6th November in Nature [...]

This is the first week of our lab at the IRB. We are excited and eager to start working here and interacting with the new colleagues. The [...]

Today is the last day of my group at UPF and the PRBB after more than 10 years. We are moving to the Barcelona Institute for [...]

The use of genomic information is becoming a key piece of the oncology toolkit to make informed decisions aimed to improve the management of the disease and increase the cost-effectiveness of available therapies.
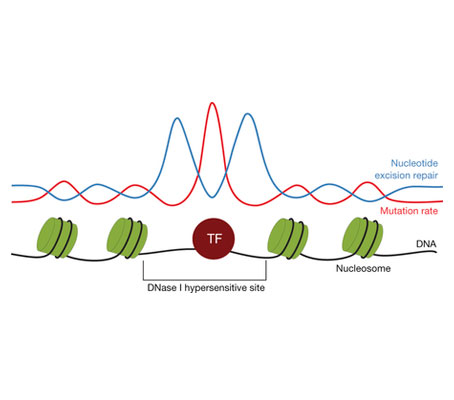
I am very happy to announce that our manuscript describing the increased mutation rate in Transcription Factor Binding Sites (TFBS) in melanomas and lung tumors has been published in today's issue of Nature. In the manuscript we demonstrate that this accumulation is due to the impairment of Nucleotide Excision Repair (NER) activity by proteins bound to DNA.
We are happy to announce that our lab has been awarded an European Research Council Consolidator Grant. ERC-consolidator grants are designed “To fund top researchers of any nationality and [...]

Coinciding with the publication of our latest paper, on Monday (9 March 2015) (See blog post) we have crafted a new IntOGen interface which presents the results of [...]
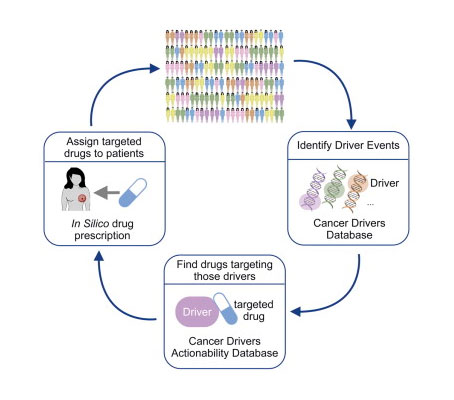
We are pleased to announce the publication of our paper in Cancer Cell describing the landscape of anti-cancer targeted therapeutic opportunities across a cohort of patients of [...]
© 2023 BBGLab