News

February 2, 2024
Barcelona Biomed Conference on CANCER PROMOTION
CANCER PROMOTION: understanding cancer promotion to inform prevention Nuria Lopez-Bigas, jointly with [...]

June 2, 2023
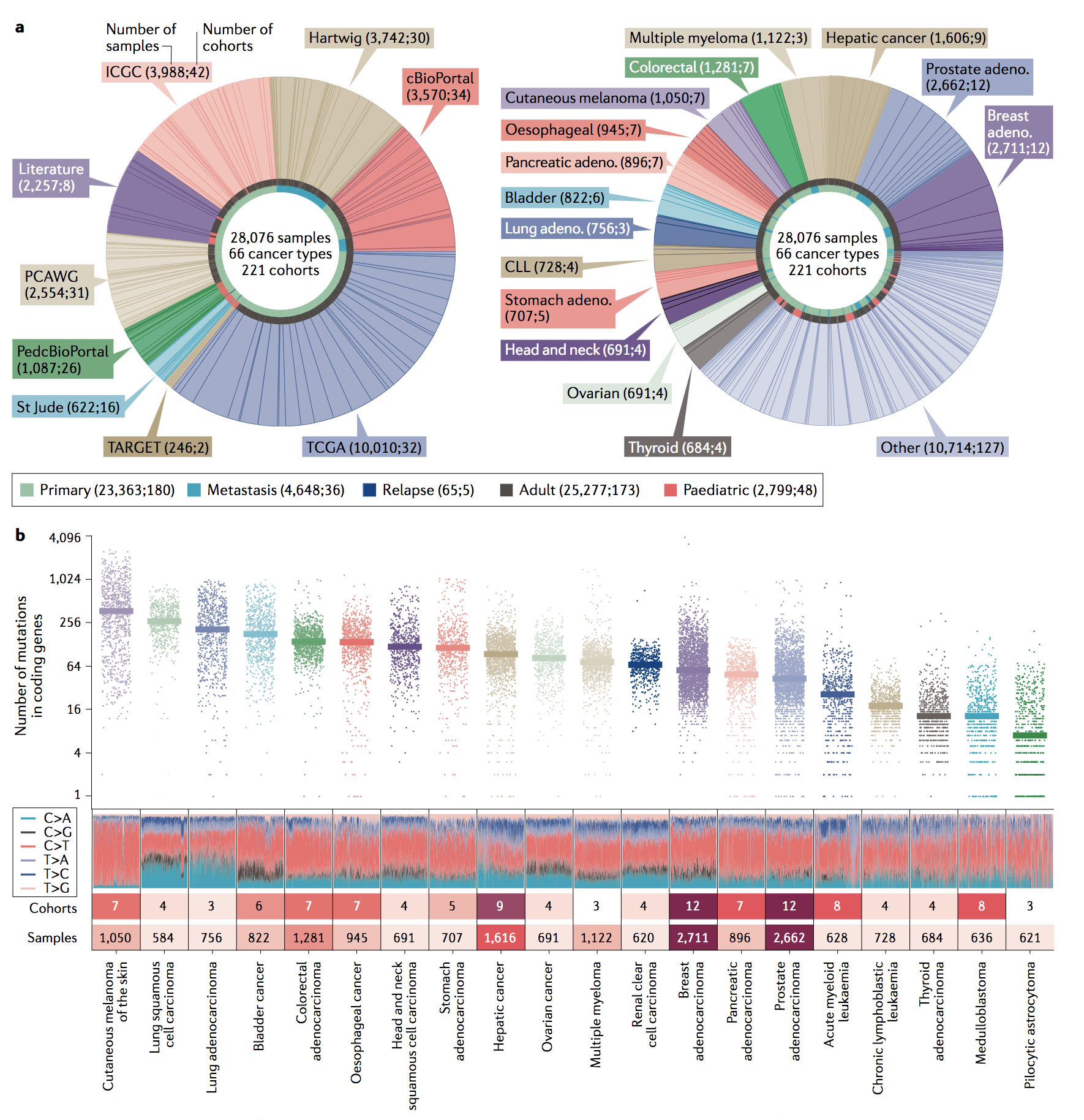
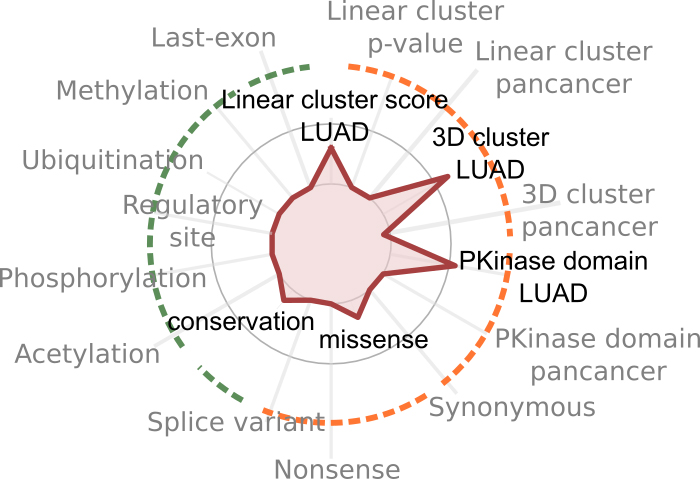
IntOGen new release (2023)
intOGen is out. In this release we have increased the number of [...]